Fall Frühjahr 2026
Beschreibung:
Eine jetzt 62-jährige Patientin mit Zustand nach lange bekanntem Mamma-Ca kommt in die Notaufnahme. Die Patientin erlitt akut ein Femurfraktur links ohne adäquates Trauma. Welche Erklärung haben Sie für die Femurfraktur und evtl. weitere im Thorax-Abdomen-CT sichtbare Knochenveränderungen? Nennen Sie bitte nur eine, die Ihnen wahrscheinlichste, Ursache.
Zur Beurteilung liegen vor:
Aktuelles Thorax-Abdomen CT
Wir benutzen zur Präsentation der DICOM Bilder den Berlin Case Viewer und danken Prof Dr. Kay-Geert Hermann, Berlin für die Überlassung.
Pathologische subtrochantäre Femurfraktur links und „frozen bone“ nach langjähriger Bisphosphonattherapie
Nebenbefundlich: Skelettmetastase im rechten prox. Femur (zweimal bioptisch negativ)
Dr. Tina Blunck, Karlsruhe
hat die Diagnose korrekt gestellt und wurde aus den richtigen Einsendungen ausgelost. Herzlichen Glückwunsch!
Wir verzeichneten 19 Einsendungen mit 15(!) richtigen Diagnosen. Respekt!
Der Fall ist spannend und wird hier im Detail vorgestellt.
Wir stellten Ihnen folgende Frage:
„Eine jetzt 62-jährige Patientin mit Zustand nach lange bekanntem Mamma-Ca kommt in die Notaufnahme. Die Patientin erlitt akut ein Femurfraktur links ohne adäquates Trauma. Welche Erklärung haben Sie für die Femurfraktur und evtl. weitere im Thorax-Abdomen-CT sichtbare Knochenveränderungen? Nennen Sie bitte nur eine, die Ihnen wahrscheinlichste, Ursache.“
Dieser Fall ist ungewöhnlich.
- Die große Mehrzahl der Kolleginnen und Kolleginnen haben die Diagnose bzw. die Ursache der Fraktur richtig erkannt. Das gab es in unserer langjährigen Geschichte des „aktuellen Falls“ noch nie.
- Die osteoplastische Metastase wurde in den Einsendungen nur vereinzelt erwähnt, was nicht verwunderlich ist, da nur nach „der Ursache“ der pathologischen subtrochantären Fraktur gefragt und Mehrfachnennungen nicht erbeten waren. Diese Metastase ist in unserem Fall eine „eigene Geschichte“…
Wir starten mit der Befundung des Thorax-Abdomen-CTs, zum Zeitpunkt nach Diagnose der subtrochantären Femurfraktur links.
Die Untersuchng wurde als orientierende „Staging“ Untersuchung konzipiert, mit der Frage nach Organmetastasen. Diese konnten verneint werden. Wir haben uns bei den von uns zur Verfügung getellten CT-Sequenzen auf den Knochen fokussiert.
Thorax-/Abdomen-CT vom Juli 2025 (MDCT mit iv.-KM-Gabe vom Lungenapex bis zur Symphyse; ergänzt durch ein natives Becken-CT mit prox. Oberschenkel 7/2025:
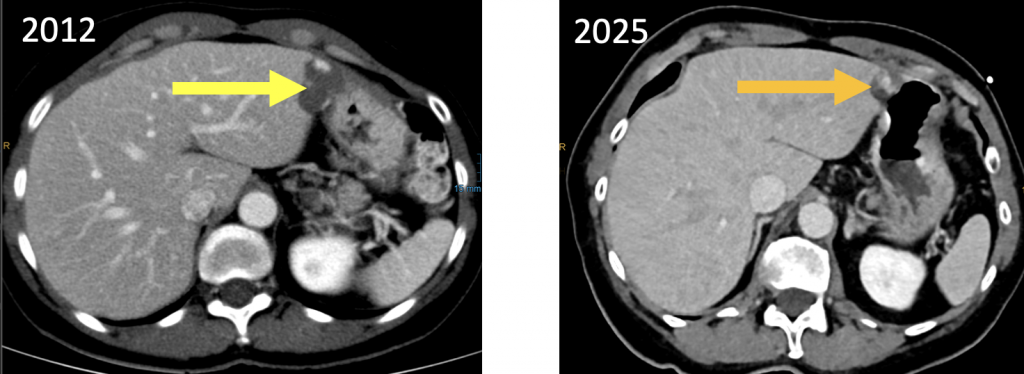
Hinsichtlich von Organmetastasen bzw. pathologisches Lymphknoten bot die Computertomographie keinen Hinweis auf die Existenz derartiger Veränderungen. Die von einem Quiz-Teilnehmer gemutmaßte Angiosarkom-verdächtige Leberläsion im medialen Randbereich des linken Leberlappen Segment S2 kann dank Verlaufsbeobachtung eindeutig einem benignen Befund zugeordnet werden: die Läsion war bereits 13 Jahre zuvor (2012), sogar noch größer, erkennbar und zeigt damals ein typisches cotton-wool-enhancement Muster, welches aktuell zwar auch noch, nur kleiner erkennbar ist; zudem enthält die Läsion eindeutig Makrofett. Es handelt sich also um ein im Langzeitverlauf größenregredientes (degenerierendes) Leberhämangiom. (Abb. 0)
|
Abb. 0. zwei KM-unterstützte Abdomen-CT´s im Abstand von 13 Jahren (differente KM-Phasen): die größenregrediente Läsion in S2 des linken Leberlappens entspricht einem typischen Leberhämangiom. Die fettäquidensen Anteile sind hier nicht gezeigt. Kein Angiosarkom o.ä. |
Zu den Knochenbefunden in beiden CTs:
1.
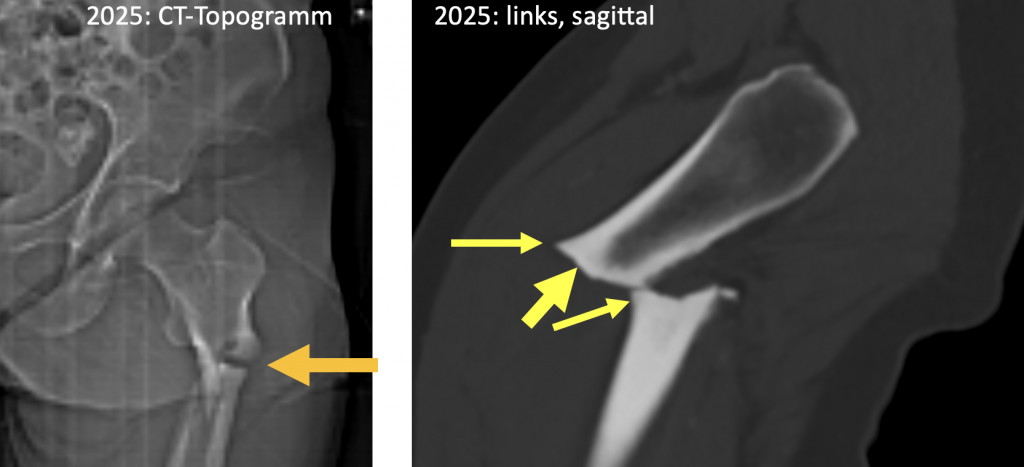
Im nativen Becken-CT am auffälligsten ist die sog. atypische proximale Femurfraktur (terminus technicus) links. Atypisch deswegen, weil die genaue Betrachtung dieser Fraktur einige Eigentümlichkeiten aufzeigt: ihr lateraler Anteil verläuft ungewöhnlich glatt und horizontal, während ihr medialer Anteil spornartig nach distal ausbricht. Zudem erkennt man im lateralen Anteil der Fraktur eine wulstartige Verbreiterung der Kompakta (beaking / flaring sign), welche medial fehlt. Isolierte Frakturfragmente sieht man nicht. Sie unterscheidet sich damit stark von den typischen Spiral- und Mehrfragmentfrakturen nach Rasanztraumen. (Abb. 1)
|
Abb. 1) Subtrochantäre atypische Femurfraktur links 2025 (oranger Pfeil): Man beachte die wulst- bzw. spornartige Kompaktaverdickung anterolateral (schmale gelbe Pfeile) sowie den zunächst horizontal gestellten Frakturverlauf (dicker gelber Pfeil) |
2.
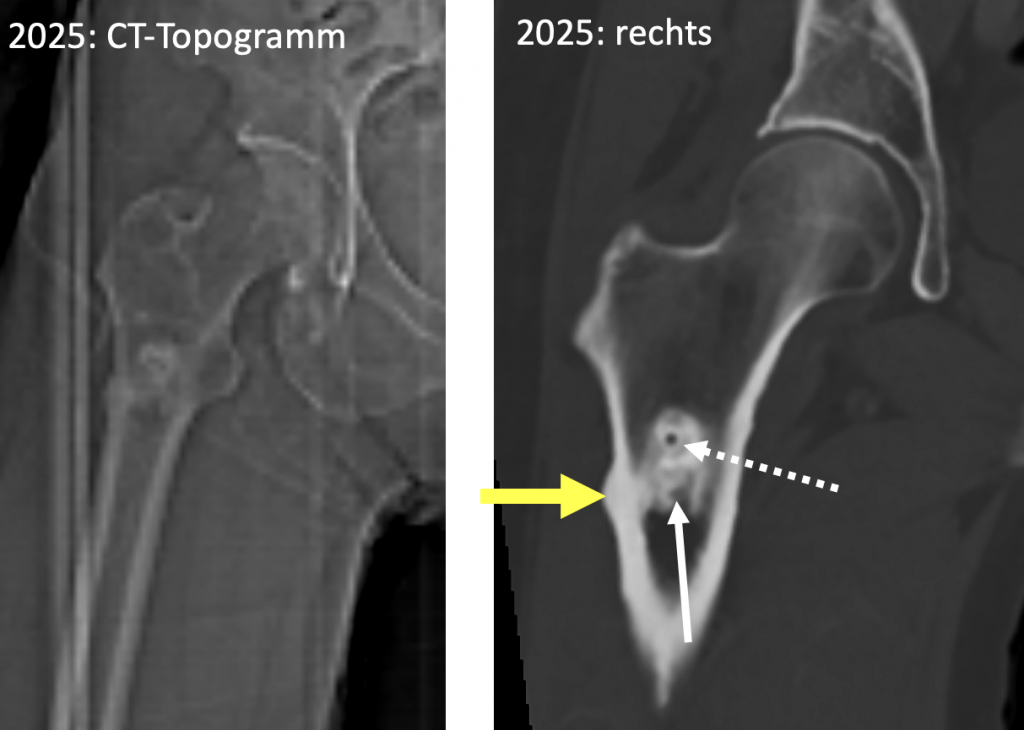
Auf der rechten Seite fällt auf gleicher Höhe wie links eine fokale, wulstartige Verbreiterung der lateralen Femurkompakta auf, nur die Fraktur(linie selbst fehlt hier (also keine sog. dreaded black line). Es handelt sich um eine typische „Vorstufe“ einer inzipienten atypischen Femurfraktur. (Abb. 2)
|
Abb. 2) inzipiente subtrochantäre atypische Femurstressreaktion rechts lateral: kortikale Wulstung (gelber Pfeil), aber noch keine Frakturlinie (inzipiente Insuff.-fraktur!). Irregulär konturierte medulläre osteosklerotische Läsion (weißer Pfeil) mit persistierendem Biopsiebohrkanal (gestrichelter Pfeil) |
3.
Gleich benachbart zur kortikalen Wulstung rechts befindet sich eine runde, dabei irregulär, aber scharf begrenzte osteosklerotische Läsion im Zentrum des prox. Femurmarkraumes. Sie ist selbst etwas inhomogen, bleibt aber dabei kräftig sklerosiert, wobei ein kokardenartiges Muster auffällt mit einem dichteren Zentrum (mittl. Dichte 1.300 HE) und einer etwas flaueren Peripherie (mittl. Dichte 910 HE; umgekehrt bizonaler Aufbau). (1) Im Zentrum dieser Sklerose findet man in der koronaren Darstellung ein kleines „Loch“, dem in der axialen Schicht der längliche Biopsiebohrkanal (der zweiten Biopsie) entspricht. (Abb. 3)
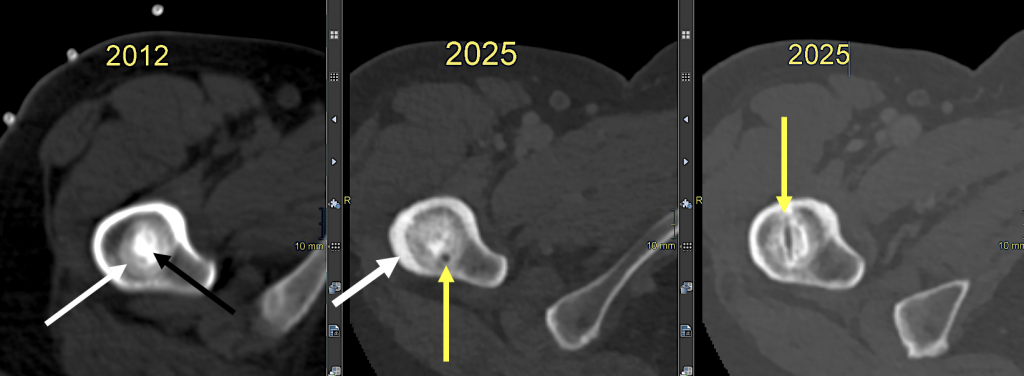
Abb. 3) 2012: Die initiale, präbioptische Darstellung der metastasensuspekten osteosklerotischen Läsion zeigt ein Target-Muster mit mäßiger peripherer Attenuierung (weißer Pfeil) und starker zentraler Sklerose (schwarzer Pfeil): sog. umgekehrt bizonaler Aufbau sklerosierter Knochenläsionen.
2025: Dreizehn Jahre später sind die Biopsiekanäle noch immer erkennbar (gelbe Pfeile). Die Läsionsgröße blieb konstant, die läsionale Dichtedifferenz hat sich verringert. Nahe der beschriebenen kortikalen Wulstung ist die Kompaktaverbreiterung bereits erkennbar (weißer Pfeil).
Diese Läsion wurde als metastasentypisch angesehen, zumal sie auch eine (allerdings geringe) FDG-Avidität aufwies. Die Läsion wurde 2012 deswegen zweimal biopsiert. (Abb. 4)
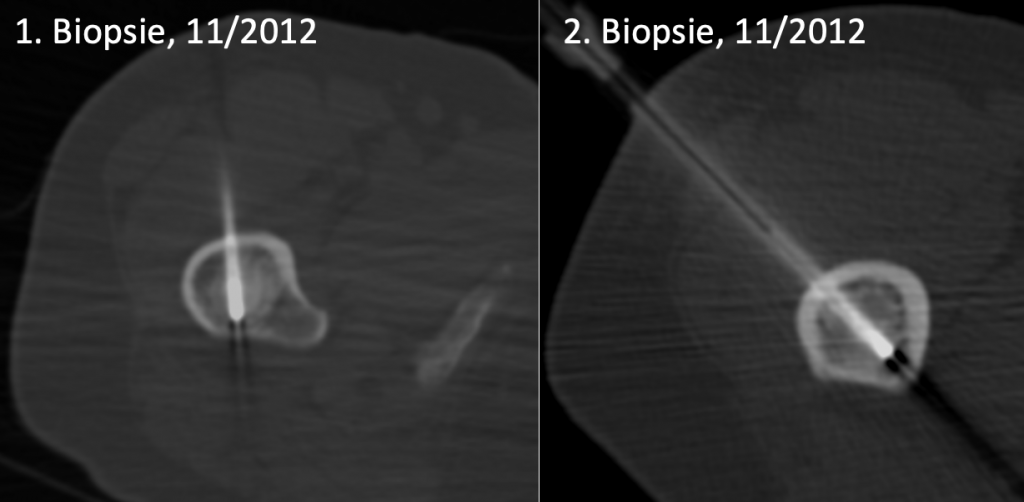
|
Abb. 4) Beide Knochenbiopsien zeigen, dass sie exakt in der Läsion lagen. Die erste Biopsie erfolge mit 11 Gauge, die zweite mit 8 Gauge. Inspektorisch wurden damals repräsentative Knochenzylinder geborgen. |
4.
Weiterhin fallen knöcherne Stressreaktionen und Insuffizienzfrakturen an den jeweiligen Pedikeln bzw. unteren Interartikulaportionen von LWK4 (symmetrische Sklerose als Folge einer Stressreaktion) bzw. LWK5 (symmetrische Insuffizienzfraktur), wobei letztere sich zu Pseudarthrosen entwickelt haben. (Abb. 5)
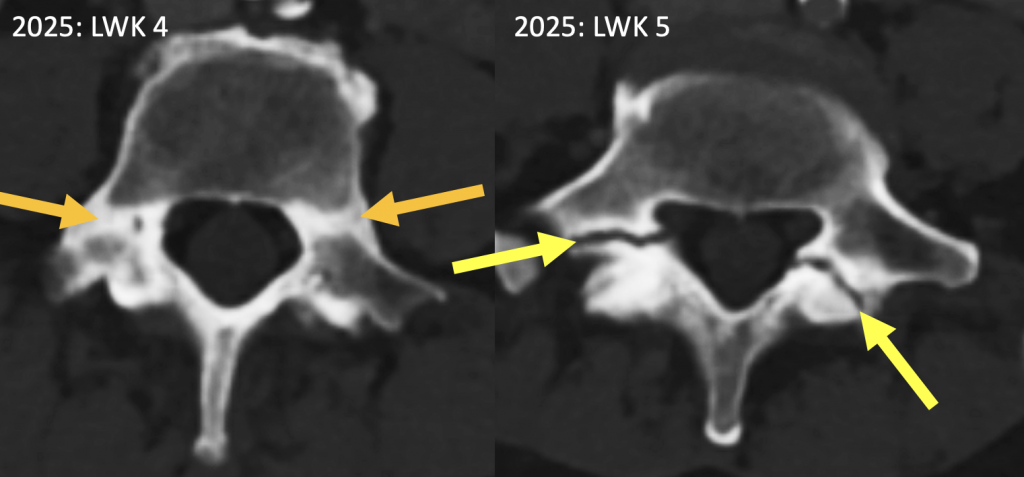
|
Abb. 5) Bandförmig absklerosierte pedikuläre Stressreaktionen (orange Pfeile, LWK 4) sowie absklerosierte Pseudarthrosen (gelbe Pfeile, LWK 5), die es alle 2012 noch nicht gab (hier nicht gezeigt). |
5.
Ganz ähnliche knöcherne Veränderungen – in unterschiedlicher Ausprägung – findet man an mehreren WS-nahen Rippenpaaren ein-, aber auch beidseitig. Entweder sieht man Rippenpseudarthrosen mit aufgeworfenen Rändern oder aber fokale kostale Mehrsklerosierungen, die sich sehr proximal und in unmittelbarer Nähe zu ihren jeweiligen kostotransversalen Gelenken befinden. Diese Regionen stellen die mechanisch am stärksten belasteten Rippenabschnitte infolge thorakaler Atemexkursionen dar. Es handelt sich also um stressinduzierte Insuffizienzreaktionen bzw. -frakturen. Vereinzelt sind auch die thorakalen Querfortsätze von den Frakturen betroffen. (Abb. 6)
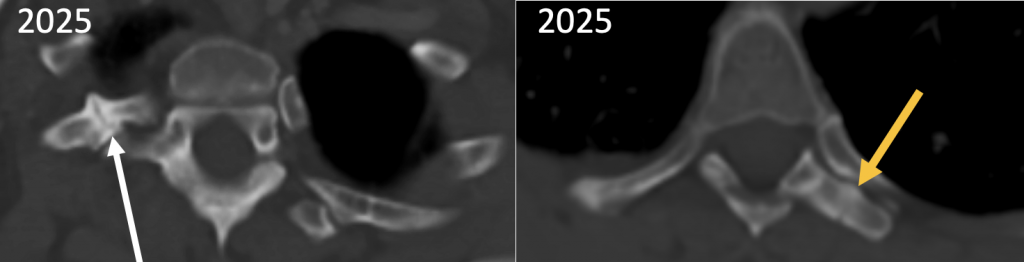
|
Abb. 6) Eine Auswahl aus den multiplen dorsalen Rippenfrakturen mit pseudarthrotischer Umwandlung (weißer Pfeil) und einer Querfortsatzfraktur (oranger Pfeil), die allesamt knochenszintigraphisch zunächst als Rippenmetastasen gewertet wurden. |
All die genannten knöchernen Veränderungen wurden zuvor szintigraphisch als Knochenmetastasen angesprochen!
Weitere Befunde
Osteonekrose des Kiefers (nicht auf diesem CT): ein auswärtiges OPG aus dem Jahr 2022 zeigte des weiteren eine Osteolyse im rechten Proc. alveolaris maxillae in der regio 24-26, eine chronische Periodontitis apicalis 26, v.a. aber eine starke Mehrsklerosierung des Alveolarkammes, vereinbar mit einer BP-induzierten Osteonekrose des Kiefers (MRONJ). Dieser Teil des Kiefers wurde 8/2022 reseziert (s. OPG 2025). (Abb. 7)
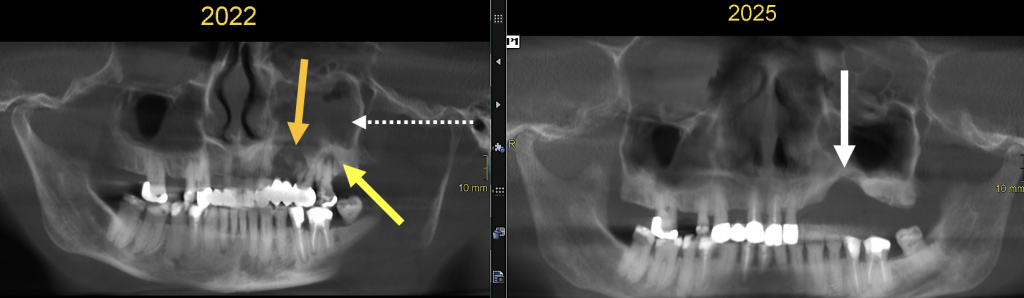
|
Abb. 7) BP-induzierte Osteonekrose im linken Oberkiefer mit entzündlicher Osteolyse (oranger Pfeil) und starker reaktiv-entzündlicher Sklerose (gelber Pfeil); zusätzlich Periodontitis apicalis 27. Drei Jahre später Zustand nach Ausräumung der Knochennekrose mit Zahnextraktionen im linken OK mit konsekutiver knöcherner Defektbildung (weißer Pfeil). Beachte die assoziierte Sinusitis maxillaris links (gestrich. Pfeil), die sich 2025 zurückgebildet hat. |
Die Krankengeschichte:
Die Patientin wurde stets auswärtig diagnostiziert und behandelt. Im Oktober 2006 wurde bei ihr ein Mammakarzinom links entdeckt, sie wurde operiert und anschließend mittels Radiochemotherapie adjuvant nachbehandelt. Dies schloß auch eine Bestrahlung des als isolierte Knochenmetastase im rechten proximalen Femur erkannten osteoplastischen Herdes (40 Gy) ein.
Seit Ende 2006 erfolgte auch eine kontinuierlich fortgesetzte, zunächst intravenöse, später orale Bisphosphonattherapie mit Zolendronat.
Warum es 2012 zur zweimaligen Biopsie der als Metastase erkannten und auch bestrahlten Knochenläsion im prox. rechten Femur kam, ist nicht mehr rekonstruierbar. Mitgeteilt werden lediglich bewegungsabhängig auftretende Schmerzen der Patientin in dieser Region.
Anläßlich der atraumatischen atypischen prox. Femurfraktur links im Juli 2025 erfolgte eine radiologische Zweitmeinung, welche die o.g. pathologischen Knochenbefunde anders interpretierte als die Erstbefunde. Erstmalig wurde dadurch der Zusammenhang aller Knochenveränderungen mit der Langzeit-BP-Therapie eindeutig hergestellt und diskutiert, und damit die knochenszintigraphisch geäußerte, multiple Metastasenhypothese verworfen!
Was ist die Folge einer langjährigen, hochdosierten Bisphosphonat-(BP) therapie?
Die eingeleitete, hochdosierte Bisphosphonattherapie (Tumordosierung) mit Zolendronat über fast 20 Jahre, mindestens aber 15 Jahre, hinweg, war notwendig und leitliniengerecht. Sie soll sowohl die Nidation von Tumorzellen im Knochen als auch die tumorbedingte osteoklastäre Knochenresorption blockieren.
Diesen gewünschten Therapieeffekten steht aber die z.T erhebliche Suppression des Knochenstoffwechsels gegenüber (Phänomen des sog. „frozen bone“); das Wechselspiel zwischen Osteoklasten und Osteoblasten wird nachhaltig gestört. Dies wiederum hat zur Folge, dass alle Reparaturmechanismen des Knochens, die infolge seiner steten physiologischen Belastung (d.h. Bewegung unter Schwerkraftbedingungen) regelhaft auftreten (sog. physiologisches Knochenremodeling) werden in ihrer Effektivität derart kompromittiert, dass normalerweise vorkommende Mikrofrakturen nicht mehr adäquat heilen können; sie akkumulieren und verstärken sich langsam, um schließlich in einer vollständigen Fraktur zu kumulieren. Es handelt sich also um klassische Insuffizienzfrakturen. (2)
Wichtig zu wissen ist, dass diese BP-induzierten Frakturen vorwiegend auf der lateralen, auf Zug beanspruchten Seite des Femurs auftreten, die aus pathophysiologischen Gründen schlechter konsolidieren (und damit wesentlich häufiger zu manifesten Frakturen führen) als ähnliche Insuffizienzereignisse auf der auf Kompression beanspruchten Innenseite des Femur.
Ähnliche Mechanismen sind auch für die nicht oder nur schlecht konsolidierenden Frakturen an den proximalen Rippen und den Pedikeln bzw. pedikelnahen Interartikularportionen der LWK4 und 5 verantwortlich: all die genannten Lokalisationen unterliegem einer vermehrten Belastung unter physiologischen Bedingungen, also einem physiologischem Stress, dem sie aber nicht widerstehen können und damit ebenfalls Insuffizienzfrakturen anheimfallen.
Wie stark der Knochenstoffwechsel gehemmt wird, zeigt sich u.a. auch an der fehlenden Rekalzifizierung der Biopsiebohrkanäle innert von fast 13 Jahren!
Biopsie einer osteoplastischen Metastase und Beurteilung des histologischen Ergebnisses. Was müssen wir beachten?
Der osteosklerotische Herd im rechten prox. Femur wurde bereits initial 2006 als Knochenmetastase interpretiert und anschließend bestrahlt, wobei hier nicht mehr bekannt ist, ob tatsächlich auch eine histologische Sicherung erfolgte.
Die Bildgebung der Läsion 2012 zum Zeitpunkt der Biopsie zeigte eine im Querschnitt targetartig konfiguerte Läsion mit einem sog. umgekehrt bizonalen Aufbau (knochendichtes Zentrum + locker verdichtete Peripherie), was tatsächlich pathomorphologisch eher einer (ehemaligen) Metastase entspricht. Die hohen nativen Dichtewerte (s. weiter oben) würden zwar gegen eine Metastase sprechen, sind aber postradiogen und unter Langzeit-BP-Therapie diagnostisch nicht verwertbar ( 1).
Im November 2012 wurde die osteoplastische Läsion im rechten proximalen Femur lege artis biopsiert; sie ergab außer einer Markraumsklerose keinen weiteren faßbaren pathologischen Befund, insbesondere keine Malignität. Daher erfolgte kurze Zeit später eine Rebiopsie; diese wurde nun auch (mehrfach) referenzpathologisch begutachtet. Wiederum zeigt sich eine Markraumsklerose ohne Nachweis maligner Zellen. Schlußendlich wurde von den Pathologen – wie immer – mehr Material gefordert, um eine „sichere“ Diagnose zu stellen.
Dieser negative histologische Befund machte zunächst etwas ratlos, allerdings wurde zumindest aus radiologischer Sicht nicht zwingend mit einer noch aviden Metastase gerechnet. Hierfür sprachen die Größenkonstanz der Läsion und die im Verlauf eher noch zunehmende posttherapeutische Sklerosierung der Läsion. Außderdem handelte es sich ja um eine bestrahlte Läsion, so daß aufgrund der Befundkonstanz nur schwerlich von einem Rezidiv ausgegangen werden konnte. Nach Diskussion mit unseren Nuklearmedizinern wurde auch die niedrige SUV-Aktivität im FDG-PET (aus 2012) als unspezifisch und nicht malignitätssuspekt eingestuft. (Abb. 8)
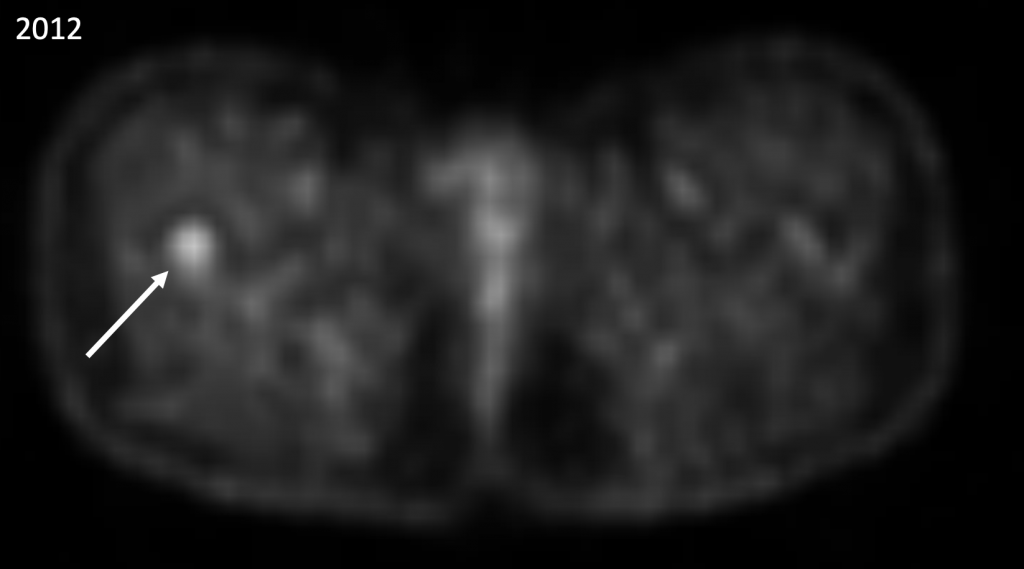
|
Abb. 8) Das FDG-PET/CT aus dem Jahre 2012 zeigt eine biologische Aktivität im Bereich der bestrahlten Femurmetastase rechts (Pfeil), was aber von unseren Nuklearmedizinern als nicht avid metastasensuspekt eingeschätzt wurde. |
Schlußendlich muß man also von einem posttherapeutisch richtig-negativen Biopsieergebnis bezüglich Malignitätsnachweis ausgehen. Der weitere Verlauf 2012 bis 2025 gibt dieser Einschätzung Recht (3).
Fazit
Der vorliegende Fall offenbart zweierlei konkrete Pathomechanismen im Rahmen der Therapie einer Krebserkrankung:
- zum einen eine schwere, therapieassoziierte und -induzierte Knochen-stoffwechselstörung, bei der es infolge der langjährigen Bisphosphonattherapie quasi zum Einfrieren des Osteometabolismus gekommen ist. Die Folge war eine pathologisch erhöhten Knochenbrüchigkeit, was sich sowohl in den manifesten und inzipienten atypischen prox. Femurfrakturen äußerte als auch in den zahlreichen Insuffizienzfrakturen an mechanisch belasteten Skelettabschnitten (Rippen, untere lumbale Pedikel) sowie in der klinisch viel schwerer wiegenden Kiefernekrose.
- Zum anderen wurde eine vorbestehende und bestrahlte Knochenmetastase als mögliches avides Rezidiv gewertet, welches durch zwei Biopsien aber nicht bestätigt werden konnte. Die Schwierigkeiten, die die Pathologen bei Beurteilung deratiger Läsionen hatten, bestätigt die diagnostische Problematik und unsichere Treffsicherheit medikamentös (Langzeit-)behandelter, bestrahlter und somit stark sklerotischer Knochenmetastasen.
Dr. Thomas Grieser
Literatur:
- Ulano A et al.: Distingushing untreated osteoblastic metastases from enostoses using CT attenuation measurements. AJR 2016; 207(2): 362-368
- Drake MT et al.: Bisphosponates: Mechanism of action and role in clinical practice. Mayo Clin Proc 2009; 83(9): 1032-1045
- Mhuicheartaigh JN et al.: Diagnostic yield of percutaneous biopsy for sclerotic bone lesions. Clin Imaging 2017; 46: 53-56.